大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全系列IDE使用 1年只要46元 售后保障 童叟无欺
遗传算法
遗传算法是用于解决最优化问题的一种搜索算法。从名字来看,遗传算法借用了生物学里达尔文的进化理论:”适者生存,不适者淘汰“,将该理论以算法的形式表现出来就是遗传算法的过程。
问题引入
上面提到遗传算法是用来解决最优化问题的,下面我将以求二元函数:
def F(x, y):
return 3*(1-x)**2*np.exp(-(x**2)-(y+1)**2)- 10*(x/5 - x**3 - y**5)*np.exp(-x**2-y**2)- 1/3**np.exp(-(x+1)**2 - y**2)
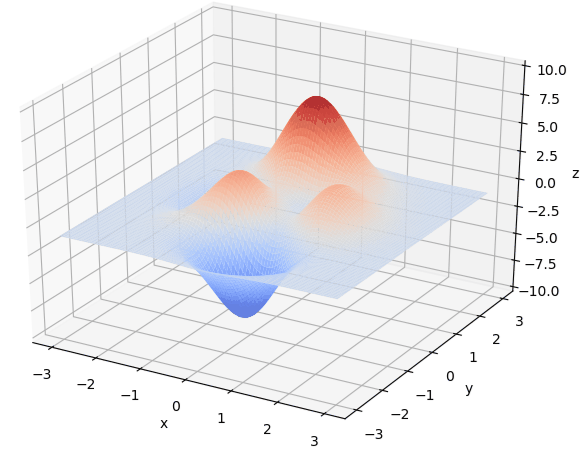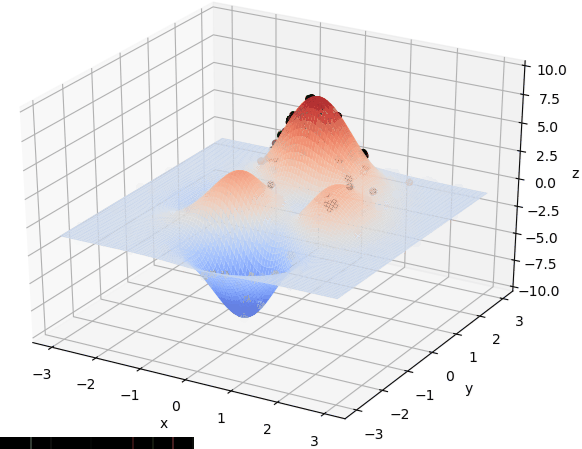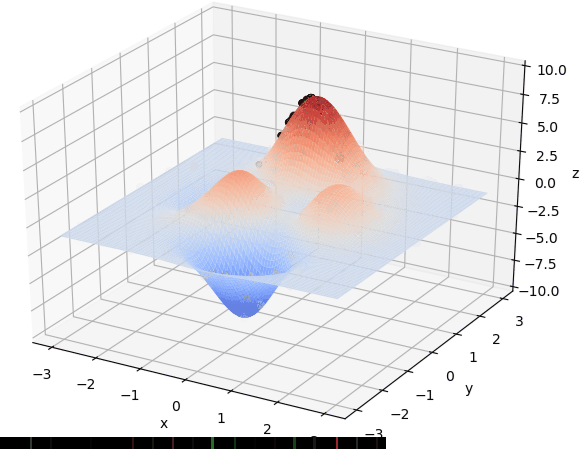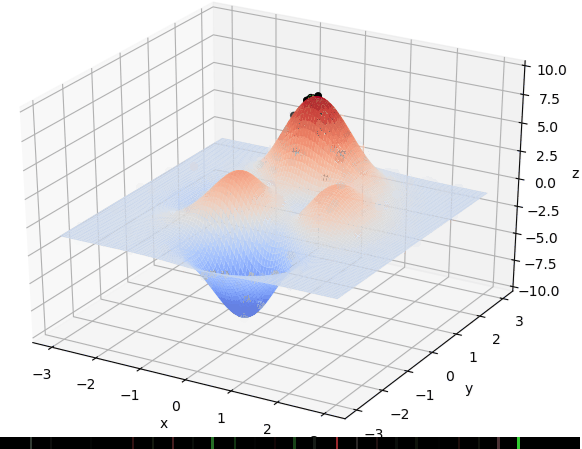
在 x ∈ [ − 3 , 3 ] , y ∈ [ − 3 , 3 ] x\in[-3, 3], y\in[-3, 3] x∈[−3,3],y∈[−3,3]范围里的最大值为例子来详细讲解遗传算法的每一步。该函数的图像如下图:
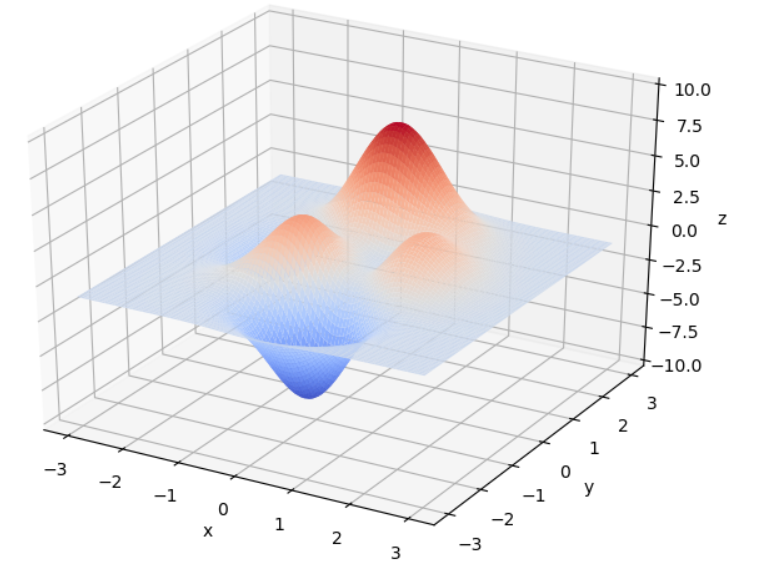

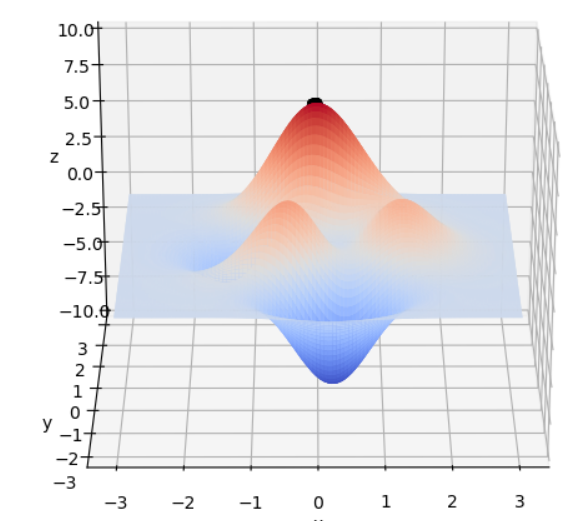
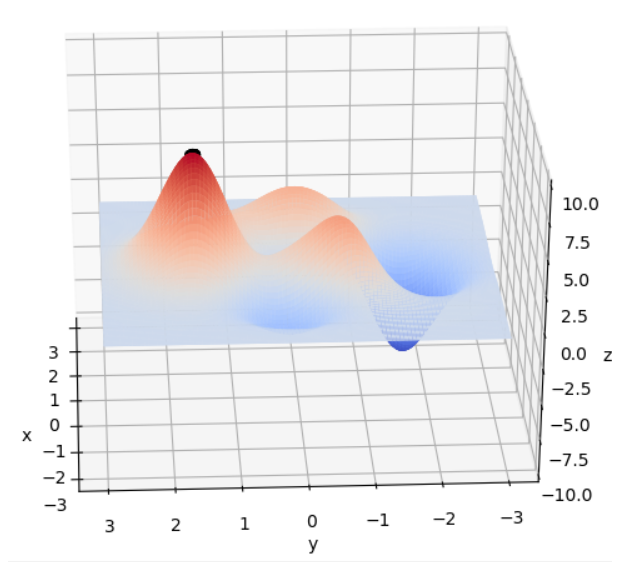
通过旋转视角可以发现,函数在这个局部的最大值大概在当 x ≈ 0 , y ≈ 1.5 x \approx 0,y\approx1.5 x≈0,y≈1.5时,函数值取得最大值,这里的 x , y x,y x,y的取值就是我们最后要得到的结果。
算法详解
先直观看一下算法过程:
寻找最小值:
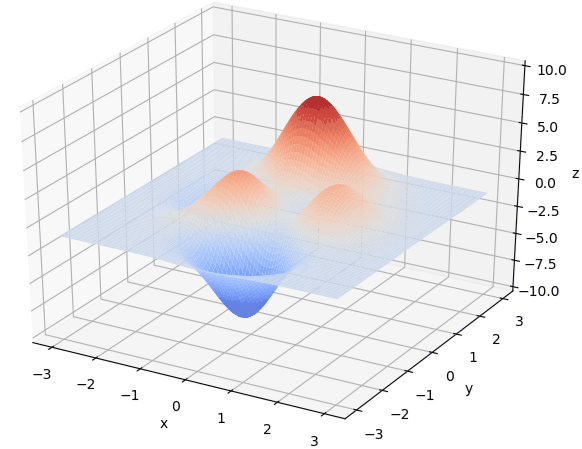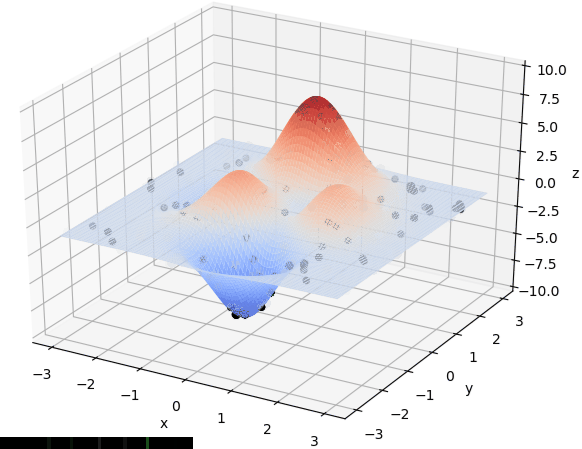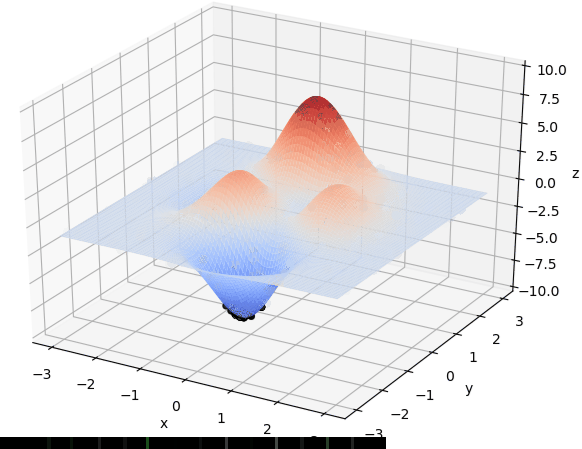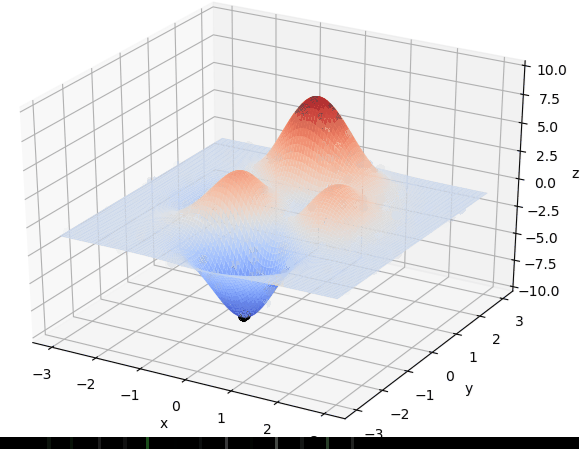
寻找最大值
首先我们生成了200个随机的(x,y)对,将(x, y)坐标对带入要求解的函数F(x,y)中,根据适者生存,我们定义使得函数值F(x,y)越大的(x,y)对越适合环境,从而这些适应环境的(x,y)对更有可能被保留下来,而那些不适应该环境的(x,y)则有很大几率被淘汰,保留下来的点经过繁殖产生新的点,如此进化下去最后留下的大部分点都是试应环境的点,即在最高点附近。下图为算法执行结果,和上面的分析 x ≈ 0 , y ≈ 1.5 x \approx 0,y\approx1.5 x≈0,y≈1.5相近。
种群和个体的概念
遗传算法启发自进化理论,而我们知道进化是由种群为单位的,种群是什么呢?维基百科上解释为:在生物学上,是在一定空间范围内同时生活着的同种生物的全部个体。显然要想理解种群的概念,又先得理解个体的概念,在遗传算法里,个体通常为某个问题的一个解,并且该解在计算机中被编码为一个向量表示! 我们的例子中要求最大值,所以该问题的解为一组可能的 ( x , y ) (x, y) (x,y)的取值。比如 ( x = 2.1 , y = 0.8 ) , ( x = − 1.5 , y = 2.3 ) . . . (x=2.1,y=0.8), (x=-1.5, y=2.3)… (x=2.1,y=0.8),(x=−1.5,y=2.3)…就是求最大值问题的一个可能解,也就是遗传算法里的个体,把这样的一组一组的可能解的集合就叫做种群 ,比如在这个问题中设置100个这样的 x , y x,y x,y的可能的取值对,这100个个体就构成了种群。
编码、解码与染色体的概念
在上面个体概念里提到个体(也就是一组可能解)在计算机程序中被编码为一个向量表示,而在我们这个问题中,个体是 x , y x,y x,y的取值,是两个实数,所以问题就可以转化为如何将实数编码为一个向量表示,可能有些朋友有疑惑,实数在计算机里不是可以直接存储吗,为什么需要编码呢?这里编码是为了后续操作(交叉和变异)的方便。实数如何编码为向量这个问题找了很多博客,写的都是很不清楚,看了莫烦python的教学代码,终于明白了一种实数编码、解码的方式。
生物的DNA有四种碱基对,分别是ACGT,DNA的编码可以看作是DNA上碱基对的不同排列,不同的排列使得基因的表现出来的性状也不同(如单眼皮双眼皮)。在计算机中,我们可以模仿这种编码,但是碱基对的种类只有两种,分别是0,1。只要我们能够将不同的实数表示成不同的0,1二进制串表示就完成了编码,也就是说其实我们并不需要去了解一个实数对应的二进制具体是多少,我们只需要保证有一个映射
y = f ( x ) , x i s d e c i m a l s y s t e m , y i s b i n a r y s y s t e m y=f(x), x \ is\ decimal \ system, y \ is \ binary\ system y=f(x),x is decimal system,y is binary system
能够将十进制的数编码为二进制即可,至于这个映射是什么,其实可以不必关心。将个体(可能解)编码后的二进制串叫做染色体,染色体(或者有人叫DNA)就是个体(可能解)的二进制编码表示。为什么可以不必关心映射 f ( x ) f(x) f(x)呢?因为其实我们在程序中操纵的都是二进制串,而二进制串生成时可以随机生成,如:
#pop表示种群矩阵,一行表示一个二进制编码表示的DNA,矩阵的行数为种群数目,DNA_SIZE为编码长度,不理解乘2的看后文
pop = np.random.randint(2, size=(POP_SIZE, DNA_SIZE*2)) #matrix (POP_SIZE, DNA_SIZE*2)
实际上是没有需求需要将一个十进制数转化为一个二进制数,而在最后我们肯定要将编码后的二进制串转换为我们理解的十进制串,所以我们需要的是 y = f ( x ) y=f(x) y=f(x)的逆映射,也就是将二进制转化为十进制,这个过程叫做解码(很重要,感觉初学者不容易理解),理解了解码编码还难吗?先看具体的解码过程如下。
首先我们限制二进制串的长度为10(长度自己指定即可,越长精度越高),例如我们有一个二进制串(在程序中用数组存储即可)
[ 0 , 1 , 0 , 1 , 1 , 1 , 0 , 1 , 0 , 1 ] [0,1,0,1,1,1,0,1,0,1] [0,1,0,1,1,1,0,1,0,1]
,这个二进制串如何转化为实数呢?不要忘记我们的 x , y ∈ [ − 3 , 3 ] x,y\in[-3,3] x,y∈[−3,3]这个限制,我们目标是求一个逆映射将这个二进制串映射到 x , y ∈ [ − 3 , 3 ] x,y\in[-3,3] x,y∈[−3,3]即可,为了更一般化我们将 x , y x,y x,y的取值范围用一个变量表示,在程序中可以用python语言写到:
X_BOUND = [-3, 3] #x取值范围
Y_BOUND = [-3, 3] #y取值范围
为将二进制串映射到指定范围,首先先将二进制串按权展开,将二进制数转化为十进制数,我们有 0 ∗ 2 9 + 1 ∗ 2 8 + 0 ∗ 2 7 + . . . + 0 ∗ 2 0 + 1 ∗ 2 0 = 373 0*2^9+1*2^8+0*2^7+…+0*2^0+1*2^0=373 0∗29+1∗28+0∗27+…+0∗20+1∗20=373,然后将转换后的实数压缩到 [ 0 , 1 ] [0,1] [0,1]之间的一个小数, 373 / ( 2 10 − 1 ) ≈ 0.36461388074 373 / (2^{10}-1) \approx 0.36461388074 373/(210−1)≈0.36461388074,通过以上这些步骤所有二进制串表示都可以转换为 [ 0 , 1 ] [0,1] [0,1]之间的小数,现在只需要将 [ 0 , 1 ] [0,1] [0,1] 区间内的数映射到我们要的区间即可。假设区间 [ 0 , 1 ] [0,1] [0,1]内的数称为num,转换在python语言中可以写成:
#X_BOUND,Y_BOUND是x,y的取值范围 X_BOUND = [-3, 3], Y_BOUND = [-3, 3],
x_ = num * (X_BOUND[1] - X_BOUND[0]) + X_BOUND[0] #映射为x范围内的数
y_ = num * (Y_BOUND[1] - Y_BOUND[0]) + Y_BOUND[0] #映射为y范围内的数
通过以上这些标记为蓝色的步骤我们完成了将一个二进制串映射到指定范围内的任务(解码)。
现在再来看看编码过程。不难看出上面我们的DNA(二进制串)长为10,10位二进制可以表示 2 10 2^{10} 210种不同的状态,可以看成是将最后要转化为的十进制区间 x , y ∈ [ − 3 , 3 ] x,y\in[-3,3] x,y∈[−3,3](下面讨论都时转化到这个区间)切分成 2 10 2^{10} 210份,显而易见,如果我们增加二进制串的长度,那么我们对区间的切分可以更加精细,转化后的十进制解也更加精确。例如,十位二进制全1按权展开为1023,最后映射到[-3, 3]区间时为3,而1111111110(前面9个1)按权展开为1022, 1022 / ( 2 10 − 1 ) ≈ 0.999022 1022/(2^{10}-1) \approx 0.999022 1022/(210−1)≈0.999022, 0.999022 ∗ ( 3 − ( − 3 ) ) + ( − 3 ) ≈ 2.994134 0.999022*(3 – (-3)) + (-3)\approx2.994134 0.999022∗(3−(−3))+(−3)≈2.994134;如果我们将实数编码为12位二进制,111111111111(12个1)最后转化为3,而111111111110(前面11个1)按权展开为4094, 4094 / ( 2 12 − 1 = 4095 ) ≈ 0.999756 4094/(2^{12}-1=4095)\approx0.999756 4094/(212−1=4095)≈0.999756, 0.999755 ∗ ( 3 − ( − 3 ) ) + ( − 3 ) ≈ 2.998534 0.999755*(3-(-3))+(-3)\approx2.998534 0.999755∗(3−(−3))+(−3)≈2.998534;而 3 − 2.994134 = 0.005866 3-2.994134=0.005866 3−2.994134=0.005866; 3 − 2.998534 = 0.001466 3-2.998534=0.001466 3−2.998534=0.001466,可以看出用10位二进制编码划分区间后,每个二进制状态改变对应的实数大约改变0.005866,而用12位二进制编码这个数字下降到0.001466,所以DNA长度越长,解码为10进制的实数越精确。
以下为解码过程的python代码:
这里我设置DNA_SIZE=24(一个实数DNA长度),两个实数 x , y x,y x,y一共用48位二进制编码,我同时将x和y编码到同一个48位的二进制串里,每一个变量用24位表示,奇数24列为x的编码表示,偶数24列为y的编码表示。
def translateDNA(pop):#pop表示种群矩阵,一行表示一个二进制编码表示的DNA,矩阵的行数为种群数目
x_pop = pop[:,1::2]#奇数列表示X
y_pop = pop[:,::2] #偶数列表示y
#pop:(POP_SIZE,DNA_SIZE)*(DNA_SIZE,1) --> (POP_SIZE,1)完成解码
x = x_pop.dot(2**np.arange(DNA_SIZE)[::-1])/float(2**DNA_SIZE-1)*(X_BOUND[1]-X_BOUND[0])+X_BOUND[0]
y = y_pop.dot(2**np.arange(DNA_SIZE)[::-1])/float(2**DNA_SIZE-1)*(Y_BOUND[1]-Y_BOUND[0])+Y_BOUND[0]
return x,y
适应度和选择
我们已经得到了一个种群,现在要根据适者生存规则把优秀的个体保存下来,同时淘汰掉那些不适应环境的个体。现在摆在我们面前的问题是如何评价一个个体对环境的适应度?在我们的求最大值的问题中可以直接用可能解(个体)对应的函数的函数值的大小来评估,这样可能解对应的函数值越大越有可能被保留下来,以求解上面定义的函数F的最大值为例,python代码如下:
def get_fitness(pop):
x,y = translateDNA(pop)
pred = F(x, y)
return (pred - np.min(pred)) + 1e-3 #减去最小的适应度是为了防止适应度出现负数,通过这一步fitness的范围为[0, np.max(pred)-np.min(pred)],最后在加上一个很小的数防止出现为0的适应度
pred是将可能解带入函数F中得到的预测值,因为后面的选择过程需要根据个体适应度确定每个个体被保留下来的概率,而概率不能是负值,所以减去预测中的最小值把适应度值的最小区间提升到从0开始,但是如果适应度为0,其对应的概率也为0,表示该个体不可能在选择中保留下来,这不符合算法思想,遗传算法不绝对否定谁也不绝对肯定谁,所以最后加上了一个很小的正数。
有了求最大值的适应度函数求最小值适应度函数也就容易了,python代码如下:
def get_fitness(pop):
x,y = translateDNA(pop)
pred = F(x, y)
return -(pred - np.max(pred)) + 1e-3
因为根据适者生存规则在求最小值问题上,函数值越小的可能解对应的适应度应该越大,同时适应度也不能为负值,先将适应度减去最大预测值,将适应度可能取值区间压缩为 [ n p . m i n ( p r e d ) − n p . m a x ( p r e d ) , 0 ] [np.min(pred)-np.max(pred), 0] [np.min(pred)−np.max(pred),0],然后添加个负号将适应度变为正数,同理为了不出现0,最后在加上一个很小的正数。
有了评估的适应度函数,下面可以根据适者生存法则将优秀者保留下来了。选择则是根据新个体的适应度进行,但同时不意味着完全以适应度高低为导向(选择top k个适应度最高的个体,容易陷入局部最优解),因为单纯选择适应度高的个体将可能导致算法快速收敛到局部最优解而非全局最优解,我们称之为早熟。作为折中,遗传算法依据原则:适应度越高,被选择的机会越高,而适应度低的,被选择的机会就低。 在python中可以写做:
def select(pop, fitness): # nature selection wrt pop's fitness
idx = np.random.choice(np.arange(POP_SIZE), size=POP_SIZE, replace=True,
p=(fitness)/(fitness.sum()) )
return pop[idx]
不熟悉numpy的朋友可以查阅一下这个函数,主要是使用了choice里的最后一个参数p,参数p描述了从np.arange(POP_SIZE)里选择每一个元素的概率,概率越高约有可能被选中,最后返回被选中的个体即可。
交叉、变异
通过选择我们得到了当前看来“还不错的基因”,但是这并不是最好的基因,我们需要通过繁殖后代(包含有交叉+变异过程)来产生比当前更好的基因,但是繁殖后代并不能保证每个后代个体的基因都比上一代优秀,这时需要继续通过选择过程来让试应环境的个体保留下来,从而完成进化,不断迭代上面这个过程种群中的个体就会一步一步地进化。
具体地繁殖后代过程包括交叉和变异两步。交叉是指每一个个体是由父亲和母亲两个个体繁殖产生,子代个体的DNA(二进制串)获得了一半父亲的DNA,一半母亲的DNA,但是这里的一半并不是真正的一半,这个位置叫做交配点,是随机产生的,可以是染色体的任意位置。通过交叉子代获得了一半来自父亲一半来自母亲的DNA,但是子代自身可能发生变异,使得其DNA即不来自父亲,也不来自母亲,在某个位置上发生随机改变,通常就是改变DNA的一个二进制位(0变到1,或者1变到0)。
需要说明的是交叉和变异不是必然发生,而是有一定概率发生。先考虑交叉,最坏情况,交叉产生的子代的DNA都比父代要差(这样算法有可能朝着优化的反方向进行,不收敛),如果交叉是有一定概率不发生,那么就能保证子代有一部分基因和当前这一代基因水平一样;而变异本质上是让算法跳出局部最优解,如果变异时常发生,或发生概率太大,那么算法到了最优解时还会不稳定。交叉概率,范围一般是0.6~1,突变常数(又称为变异概率),通常是0.1或者更小。
python实现如下:
def crossover_and_mutation(pop, CROSSOVER_RATE = 0.8):
new_pop = []
for father in pop: #遍历种群中的每一个个体,将该个体作为父亲
child = father #孩子先得到父亲的全部基因(这里我把一串二进制串的那些0,1称为基因)
if np.random.rand() < CROSSOVER_RATE: #产生子代时不是必然发生交叉,而是以一定的概率发生交叉
mother = pop[np.random.randint(POP_SIZE)] #再种群中选择另一个个体,并将该个体作为母亲
cross_points = np.random.randint(low=0, high=DNA_SIZE*2) #随机产生交叉的点
child[cross_points:] = mother[cross_points:] #孩子得到位于交叉点后的母亲的基因
mutation(child) #每个后代有一定的机率发生变异
new_pop.append(child)
return new_pop
def mutation(child, MUTATION_RATE=0.003):
if np.random.rand() < MUTATION_RATE: #以MUTATION_RATE的概率进行变异
mutate_point = np.random.randint(0, DNA_SIZE) #随机产生一个实数,代表要变异基因的位置
child[mutate_point] = child[mutate_point]^1 #将变异点的二进制为反转
上面这些步骤即为遗传算法的核心模块,将这些模块在主函数中迭代起来,让种群去进化
pop = np.random.randint(2, size=(POP_SIZE, DNA_SIZE*2)) #生成种群 matrix (POP_SIZE, DNA_SIZE)
for _ in range(N_GENERATIONS): #种群迭代进化N_GENERATIONS代
crossover_and_mutation(pop, CROSSOVER_RATE) #种群通过交叉变异产生后代
fitness = get_fitness(pop) #对种群中的每个个体进行评估
pop = select(pop, fitness) #选择生成新的种群
附录
完整代码
import numpy as np
import matplotlib.pyplot as plt
from matplotlib import cm
from mpl_toolkits.mplot3d import Axes3D
DNA_SIZE = 24
POP_SIZE = 200
CROSSOVER_RATE = 0.8
MUTATION_RATE = 0.005
N_GENERATIONS = 50
X_BOUND = [-3, 3]
Y_BOUND = [-3, 3]
def F(x, y):
return 3*(1-x)**2*np.exp(-(x**2)-(y+1)**2)- 10*(x/5 - x**3 - y**5)*np.exp(-x**2-y**2)- 1/3**np.exp(-(x+1)**2 - y**2)
def plot_3d(ax):
X = np.linspace(*X_BOUND, 100)
Y = np.linspace(*Y_BOUND, 100)
X,Y = np.meshgrid(X, Y)
Z = F(X, Y)
ax.plot_surface(X,Y,Z,rstride=1,cstride=1,cmap=cm.coolwarm)
ax.set_zlim(-10,10)
ax.set_xlabel('x')
ax.set_ylabel('y')
ax.set_zlabel('z')
plt.pause(3)
plt.show()
def get_fitness(pop):
x,y = translateDNA(pop)
pred = F(x, y)
return (pred - np.min(pred)) + 1e-3 #减去最小的适应度是为了防止适应度出现负数,通过这一步fitness的范围为[0, np.max(pred)-np.min(pred)],最后在加上一个很小的数防止出现为0的适应度
def translateDNA(pop): #pop表示种群矩阵,一行表示一个二进制编码表示的DNA,矩阵的行数为种群数目
x_pop = pop[:,1::2]#奇数列表示X
y_pop = pop[:,::2] #偶数列表示y
#pop:(POP_SIZE,DNA_SIZE)*(DNA_SIZE,1) --> (POP_SIZE,1)
x = x_pop.dot(2**np.arange(DNA_SIZE)[::-1])/float(2**DNA_SIZE-1)*(X_BOUND[1]-X_BOUND[0])+X_BOUND[0]
y = y_pop.dot(2**np.arange(DNA_SIZE)[::-1])/float(2**DNA_SIZE-1)*(Y_BOUND[1]-Y_BOUND[0])+Y_BOUND[0]
return x,y
def crossover_and_mutation(pop, CROSSOVER_RATE = 0.8):
new_pop = []
for father in pop: #遍历种群中的每一个个体,将该个体作为父亲
child = father #孩子先得到父亲的全部基因(这里我把一串二进制串的那些0,1称为基因)
if np.random.rand() < CROSSOVER_RATE: #产生子代时不是必然发生交叉,而是以一定的概率发生交叉
mother = pop[np.random.randint(POP_SIZE)] #再种群中选择另一个个体,并将该个体作为母亲
cross_points = np.random.randint(low=0, high=DNA_SIZE*2) #随机产生交叉的点
child[cross_points:] = mother[cross_points:] #孩子得到位于交叉点后的母亲的基因
mutation(child) #每个后代有一定的机率发生变异
new_pop.append(child)
return new_pop
def mutation(child, MUTATION_RATE=0.003):
if np.random.rand() < MUTATION_RATE: #以MUTATION_RATE的概率进行变异
mutate_point = np.random.randint(0, DNA_SIZE*2) #随机产生一个实数,代表要变异基因的位置
child[mutate_point] = child[mutate_point]^1 #将变异点的二进制为反转
def select(pop, fitness): # nature selection wrt pop's fitness
idx = np.random.choice(np.arange(POP_SIZE), size=POP_SIZE, replace=True,
p=(fitness)/(fitness.sum()) )
return pop[idx]
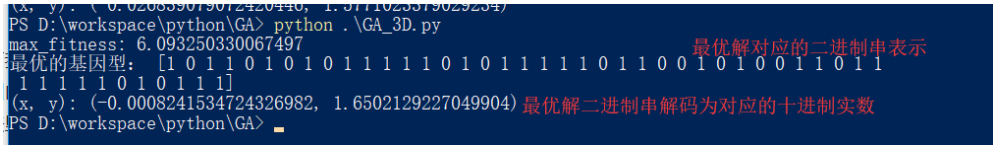
def print_info(pop):
fitness = get_fitness(pop)
max_fitness_index = np.argmax(fitness)
print("max_fitness:", fitness[max_fitness_index])
x,y = translateDNA(pop)
print("最优的基因型:", pop[max_fitness_index])
print("(x, y):", (x[max_fitness_index], y[max_fitness_index]))
if __name__ == "__main__":
fig = plt.figure()
ax = Axes3D(fig)
plt.ion()#将画图模式改为交互模式,程序遇到plt.show不会暂停,而是继续执行
plot_3d(ax)
pop = np.random.randint(2, size=(POP_SIZE, DNA_SIZE*2)) #matrix (POP_SIZE, DNA_SIZE)
for _ in range(N_GENERATIONS):#迭代N代
x,y = translateDNA(pop)
if 'sca' in locals():
sca.remove()
sca = ax.scatter(x, y, F(x,y), c='black', marker='o');plt.show();plt.pause(0.1)
pop = np.array(crossover_and_mutation(pop, CROSSOVER_RATE))
#F_values = F(translateDNA(pop)[0], translateDNA(pop)[1])#x, y --> Z matrix
fitness = get_fitness(pop)
pop = select(pop, fitness) #选择生成新的种群
print_info(pop)
plt.ioff()
plot_3d(ax)
2022年08月12日18:41:58
Update
后记:很多人评论最后最大适应度很小是不是没收敛,我说一下我的理解,请注意最大适应度和F(x,y)不等价,最大适应度小不能说明没有拟合,认真观察适应度咋算的,是F(x,y) – min(F(x,y)),这个值小,F(x,y)的最大值和最小值很接近,方差很小,反而可能是表明收敛了
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/190459.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
