大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全系列IDE使用 1年只要46元 售后保障 童叟无欺
1、下载fastqc
wget
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip
2、解压
unzip fastqc_v0.11.5.zip
3、 给予执行权限,否则执行的时候会显示没有权限
cd FastQC
chmod 755 fastqc
4、加入到PATH
export PATH=/home/h/FastQC/:$PATH
5、测试
fastqc –help
使用例子
fastqc -o ./tmp.result/fastQC/ -t 6 ./
tmp.data/fastq/H1EScell-dnase-2014-GSE56869_20151208_SRR1248176_1.fq
# -o –outdir FastQC生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的
# -t –threads 选择程序运行的线程数,每个线程会占用250MB内存,越多越快咯
FastQC的报告介绍
整个报告分成若干个部分。合格会有个绿色的对勾,警告是个“!”,不合格是个红色的叉子
基本信息
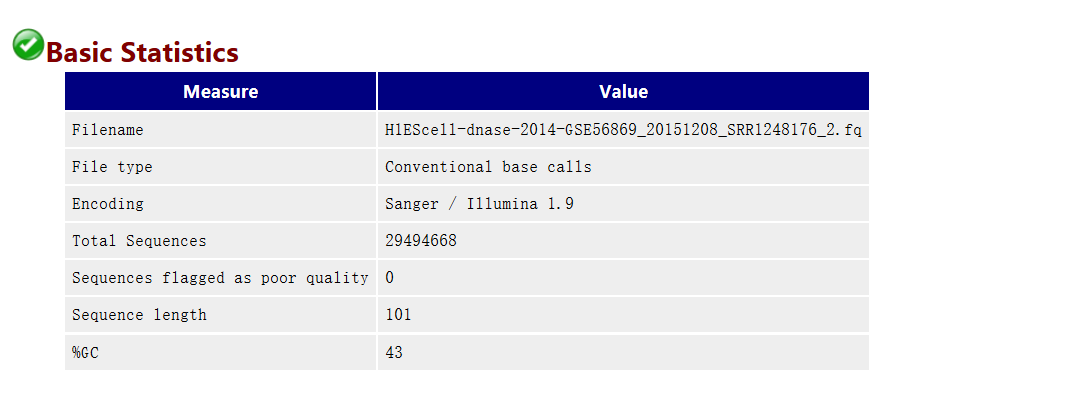

# Encoding指测序平台的版本和相应的编码版本号,这个在计算Phred反推error P的时候有用
# Total Sequences记录了输入文本的reads的数量
# Sequence length是测序的长度
# %GC是我们需要重点关注的一个指标,这个值表示的是整体序列中的GC含量,这个数值一般是物种特异的,比如人类细胞就是42%左右
序列测序质量统计
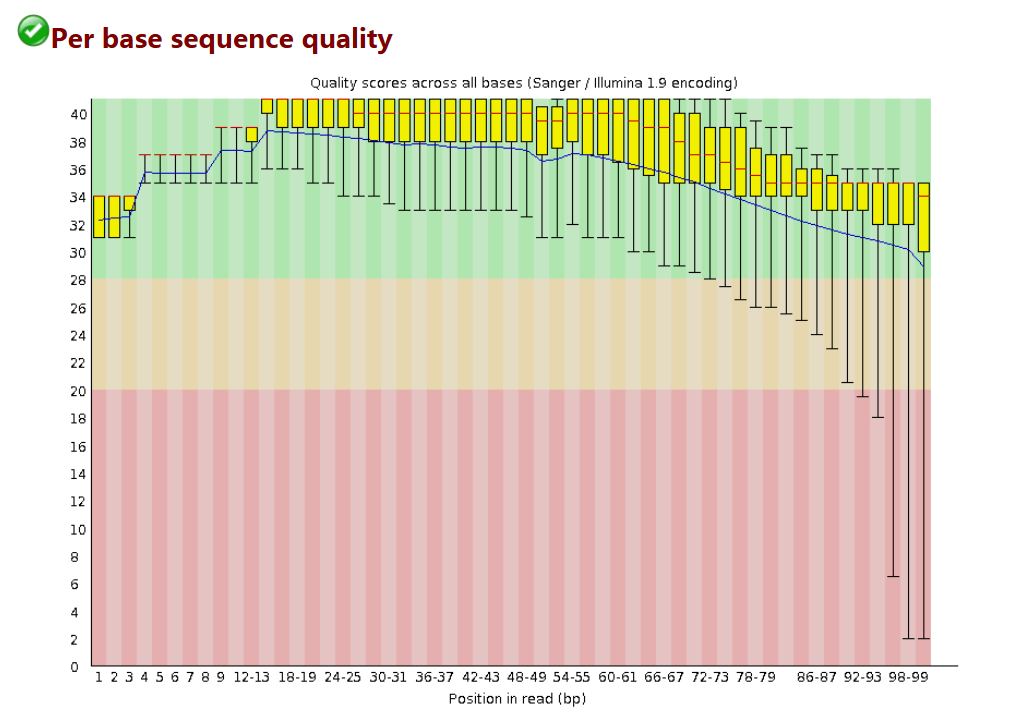
# 此图中的横轴是测序序列第1个碱基到第101个碱基
# 纵轴是质量得分,Q = -10*log10(error P)即20表示1%的错误率,30表示0.1%
# 图中每1个boxplot,都是该位置的所有序列的测序质量的一个统计,上面的bar是90%分位数,下面的bar是10%分位数,箱子的中间的横线是50%分位数,箱子的上边是75%分位数,下边是25%分位数
# 图中蓝色的细线是各个位置的平均值的连线
# 一般要求此图中,所有位置的10%分位数大于20,也就是我们常说的Q20过滤
# 所以上面的这个测序结果,需要把后面的87bp以后的序列切除,从而保证后续分析的正确性
# Warning 报警 如果任何碱基质量低于10,或者是任何中位数低于25
# Failure 报错 如果任何碱基质量低于5,或者是任何中位数低于20
每个tail测序的情况
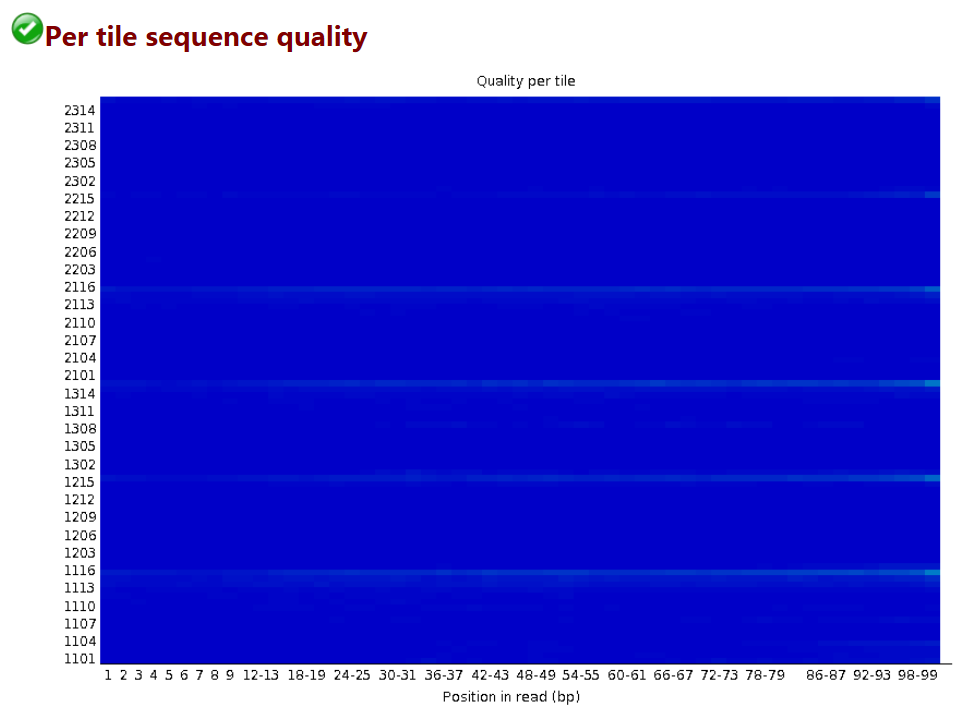
# 横轴和之前一样,代表101个碱基的每个不同位置
# 纵轴是tail的Index编号
# 这个图主要是为了防止在测序过程中,某些tail受到不可控因素的影响而出现测序质量偏低
# 蓝色代表测序质量很高,暖色代表测序质量不高,如果某些tail出现暖色,可以在后续分析中把该tail测序的结果全部都去除
每条序列的测序质量统计
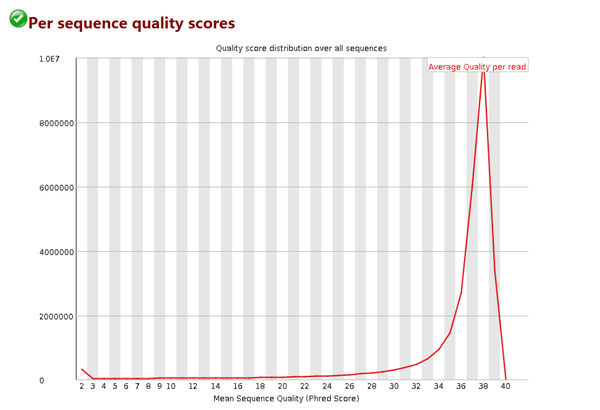
# 假如我测的1条序列长度为101bp,那么这101个位置每个位置Q值的平均值就是这条reads的质量值
# 该图横轴是0-40,表示Q值
# 纵轴是每个值对应的reads数目
# 我们的数据中,测序结果主要集中在高分中,证明测序质量良好!
GC 含量统计
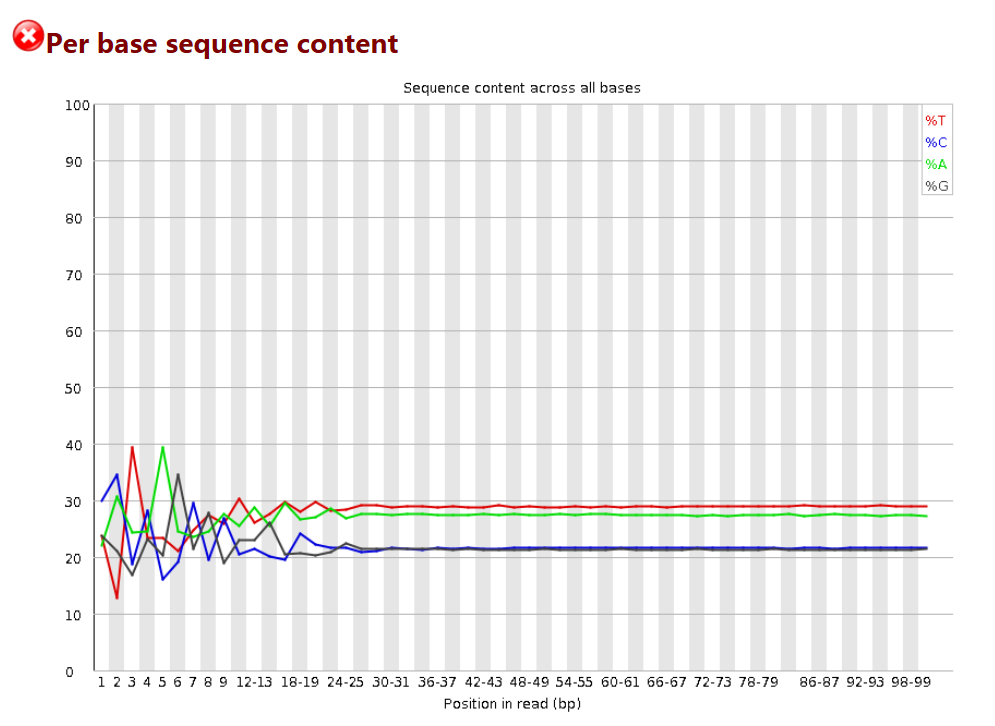
# 横轴是1 – 101 bp;纵轴是百分比
# 图中四条线代表A T C G在每个位置平均含量
# 理论上来说,A和T应该相等,G和C应该相等,但是一般测序的时候,刚开始测序仪状态不稳定,很可能出现上图的情况。像这种情况,即使测序的得分很高,也需要cut开始部分的序列信息,一般像我碰到这种情况,会cut前面5bp
序列平均GC含量分布图
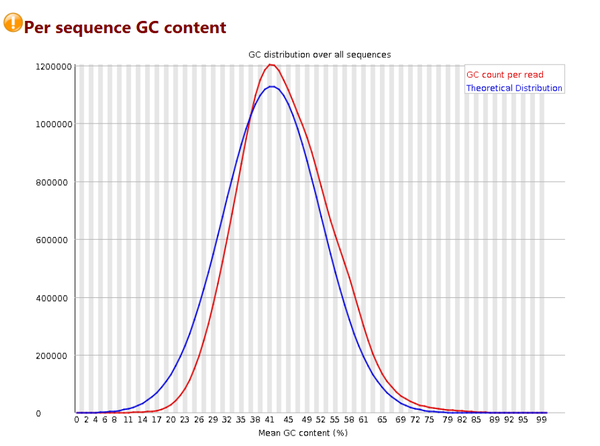
# 横轴是0 – 100%; 纵轴是每条序列GC含量对应的数量
# 蓝色的线是程序根据经验分布给出的理论值,红色是真实值,两个应该比较接近才比较好
# 当红色的线出现双峰,基本肯定是混入了其他物种的DNA序列
# 这张图中的信息良好
序列测序长度统计
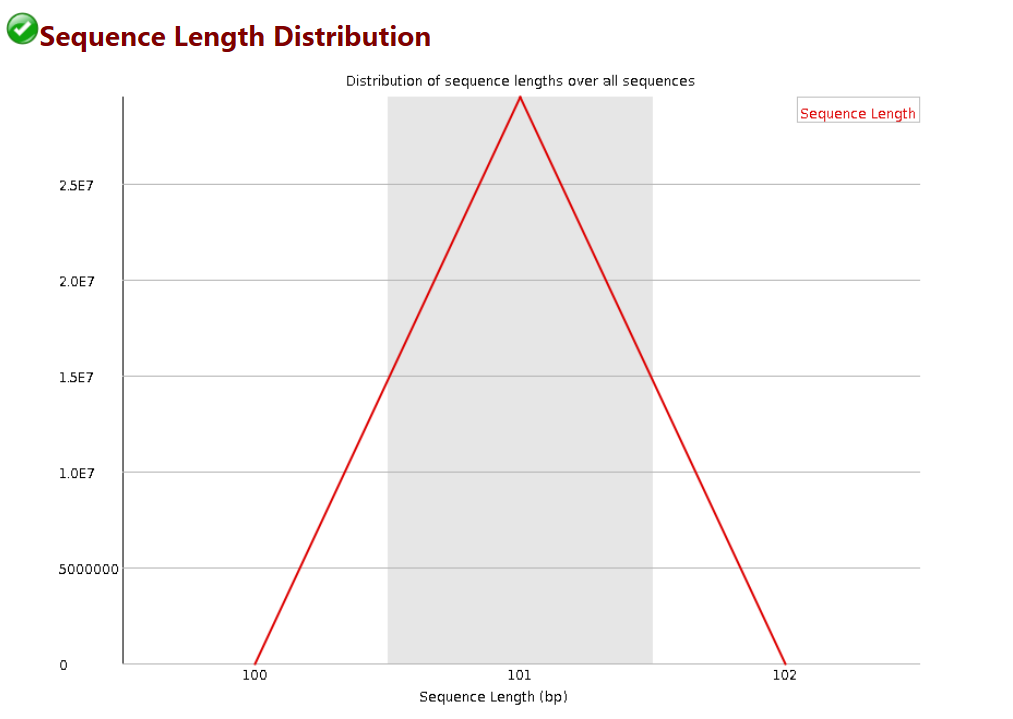
# 每次测序仪测出来的长度在理论上应该是完全相等的,但是总会有一些偏差
# 比如此图中,101bp是主要的,但是还是有少量的100和102bp的长度,不过数量比较少,不影响后续分析
# 当测序的长度不同时,如果很严重,则表明测序仪在此次测序过程中产生的数据不可信
序列Adapter
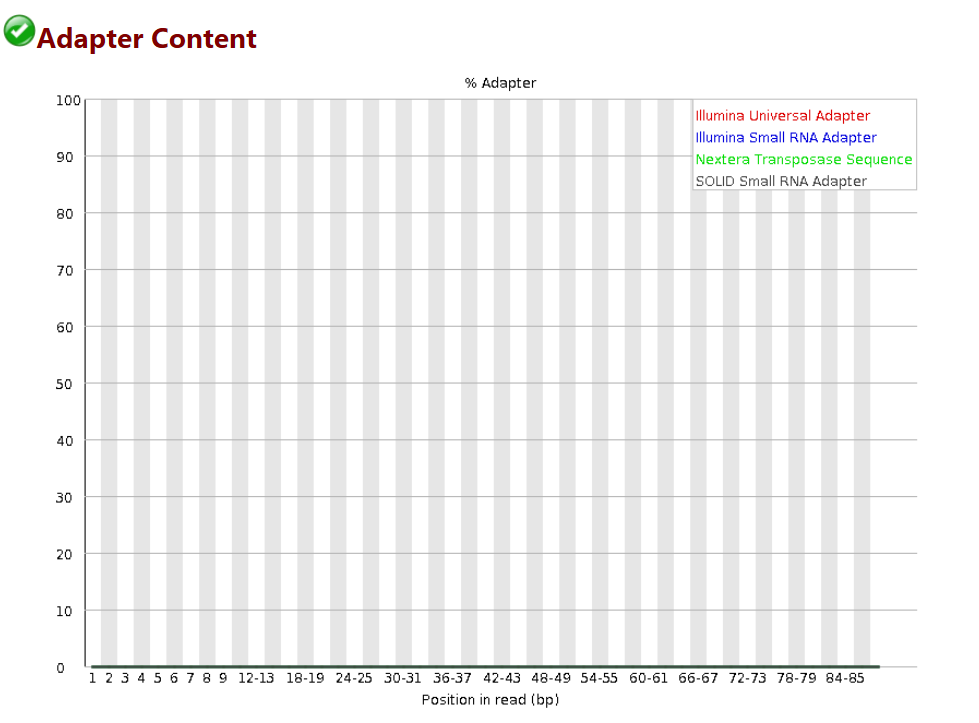
# 此图衡量的是序列中两端adapter的情况
# 如果在当时fastqc分析的时候-a选项没有内容,则默认使用图例中的四种通用adapter序列进行统计
# 本例中adapter都已经去除,如果有adapter序列没有去除干净的情况,在后续分析的时候需要先使用cutadapt软件进行去接头
重复短序列
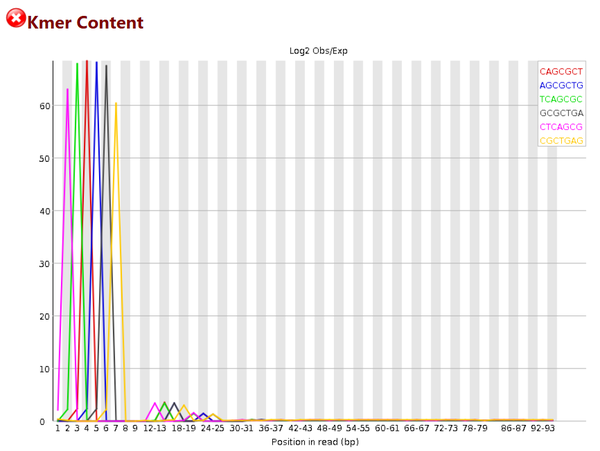
# 这个图统计的是,在序列中某些特征的短序列重复出现的次数
# 我们可以看到1-8bp的时候图例中的几种短序列都出现了非常多的次数,一般来说,出现这种情况,要么是adapter没有去除干净,而又没有使用-a参数;要么就是序列本身可能重复度比较高,如建库PCR的时候出现了bias
# 对于这种情况,我的办法是可以cut掉前面的一些长度,可以试着cut 5~8bp
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 举报,一经查实,本站将立刻删除。
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/167116.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
