大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全家桶1年46,售后保障稳定
O.Sativa选用MSU或者RAPDB这两个数据库的genome和gtf文件,介绍一下MSU的ID,RAPDB的同理。The Rice Annotation Project (RAP)(https://rapdb.dna.affrc.go.jp/index.html)和Rice Genome Annotation Project (RGAP7,MSU)(http://rice.plantbiology.msu.edu/index.shtml)RAP格式为“Os-Chr-g-number”,MSU格式为“LOC_Os-Chr-g-number”。
1 AGRIGO2 http://systemsbiology.cau.edu.cn/agriGOv2/ 支持多种ID,包括MSU
2 RIGW http://rice.hzau.edu.cn/cgi-bin/rice2/enrichment 只支持水稻的转录本ID,可做KEGG
3 PlantGSEA http://structuralbiology.cau.edu.cn/PlantGSEA/analysis.php 只支持MSU ID
4 PANTHER http://www.pantherdb.org/ 可视化漂亮。支持Uniprot ID。MSU ID转换为 Uniprot ID(PlantGSEA)
5 CARMO:http://bioinfo.sibs.ac.cn/carmo/result.php?job_id=1625924324108758969 只更新到 2015年,支持 LOC ID
- 将MSU ID(LOC)转换为 Uniprot ID,
PlantGSEA - 将Uniprot ID粘贴到
PANTHER中
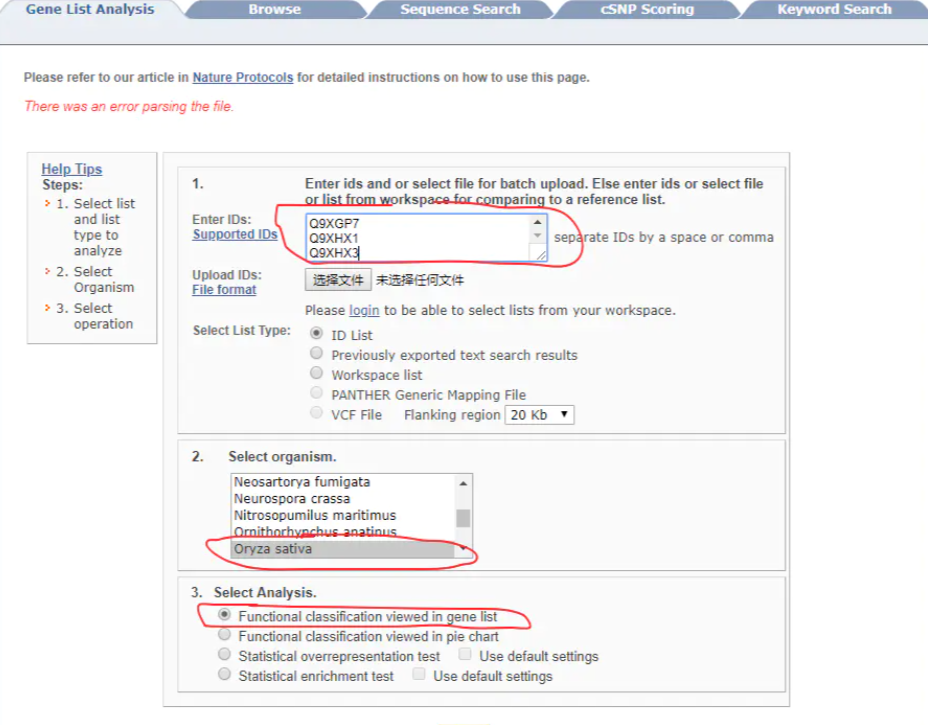

- 出图, select ontology,包括GO分析,蛋白功能注释,Pathway分析
- 显著性分析,statistical overrepresentation test, Use default settings
ID转化
- 水稻的基因号大致分为两类,RAP格式为“Os-Chr-g-number”,MSU格式为“LOC_Os-Chr-g-number”。各种分析输入的基因号有指定要求,ID转换至关重要。
- RAP《-》MSU:(OryzaExpress,RAP-DB,PlantGSEA)
OryzaExpress:http://bioinf.mind.meiji.ac.jp/OryzaExpress/ID_converter.phpRAP-DB:https://rapdb.dna.affrc.go.jp/tools/converter/runPlantGSEA:http://structuralbiology.cau.edu.cn/PlantGSEA/- Ensemble Plants(http://plants.ensembl.org/index.html),
- RIGW(http://rice.hzau.edu.cn/rice/)
biomaRt RAP转entrezgene_id(NCBI)- MSU转RAP转entrezid,MSU转uniprot(plantGSEA)转entrezid(david)
biomaRt
#1.Installation
BiocManager::install("biomaRt")
library(biomaRt)
#2.Data Import
a <- read.csv('testgene.txt',sep = '\t')
#3.getBM做ID转换
#getBM函数,四个参数。 getBM函数唯一用处,做各种ID转换。
#1.filter来控制根据什么东西来过滤,可是不同数据库的ID,也可以是染色体定位系统坐标。
#2.Attributes来控制我们想获得什么,一般是不同数据库的ID。
#3.Values是我们用来检索的关键词向量。
#4.Mart是我们前面选择好的数据库。
#3.1建立与ensemble数据库的链接
#在ensemble plants上能看到所有已提交的物种信息
ensembl = useMart(biomart = "plants_mart",host = "http://plants.ensembl.org")
#查看ensemble plants都有哪些物种信息,并设置为该物种信息。
dataset <- listDatasets(mart = ensembl)
head(dataset)
ensembl = useMart(biomart = "plants_mart",host = "http://plants.ensembl.org",dataset="osativa_eg_gene")
#查看该dataset上都有哪些属性,方便后面做添加
attributes <- listAttributes(ensembl)
a=c(“LOC_Os07g34570”,“LOC_Os05g12630”,“LOC_Os12g31000”,“LOC_Os09g37910”)无结果
RAP:
3.2 正式做ID转换及信息添加
一般Ensemble ID以E开头的,RAP号是水稻的Ensemble ID。参数external_gene_name是平时称呼的基因名字。
supplement <- getBM(attributes =c("ensembl_gene_id",'external_gene_name',"description"),filters = "ensembl_gene_id",values = a,mart = ensembl)
转换成GO ID并附上GO描述
supplements <- getBM(attributes =c("ensembl_gene_id",'go_id','goslim_goa_description'),
filters = "ensembl_gene_id",values = a,mart = ensembl)
转换成NCBI ID
supplements <- getBM(attributes =c("ensembl_gene_id",'entrezgene_id'),
filters = "ensembl_gene_id",values = a,mart = ensembl)
ClusterProfile
-
GO数据库?以及BP,MF,CC的分类系统?超几何分布检验?不同的阈值过滤?筛选指标?
-
超几何分布是统计学上一种离散概率分布。它描述了从有限N个物件(其中包含M个指定种类的物件)中抽出n个物件,成功抽出该指定种类的物件的次数(不放回)。
-
拿不到结果?这个时候可以设置: pvalueCutoff = 0.9, qvalueCutoff =0.9 甚至为1,来不做筛选。而且基因集的大小也是被限制了。
enrichGO(); dotplot() -
多组基因集的KEGG数据库富集
compareCluster,在线获取KEGG数据库最新信息,考验网速: -
制作一个 DEG 数据框,其中有两列ENTREZID,是基因id,和new是分组信息
xx.formula <- compareCluster(ENTREZID~new, data=DEG, fun=‘enrichKEGG’)
dotplot(xx.formula, x=~GeneRatio) + facet_grid(~new) -
多组基因集走GO数据库富集
构建一个数据框,list_de_gene_clusters, 含有两列信息:
list_de_gene_clusters <- split(de_gene_clusters$ENTREZID,
de_gene_clusters$cluster)
# Run full GO enrichment test
formula_res <- compareCluster(
ENTREZID~cluster,
data=de_gene_clusters,
fun="enrichGO",
OrgDb="org.Mm.eg.db",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05
)
# Run GO enrichment test and merge terms
# that are close to each other to remove result redundancy
lineage1_ego <- simplify(
formula_res,
cutoff=0.5,
by="p.adjust",
select_fun=min
)
https://www.jianshu.com/p/bcdbf80701e2
https://www.jianshu.com/p/480c46ec1629
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/227221.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
