大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全家桶1年46,售后保障稳定
1. 软件下载及安装admixture:
使用conda进行软件安装
conda install admixture2. VCF文件格式转换为bed格式文件(似乎admixture 可以直接识别ped/map文件格式的输入文件)
vcf文件转为ped文件:
方法1:
使用vcftools支持将vcf文件转换成plink对应的ped/map格式,如下
vcftools --vcf input.vcf --plink --out output方法2:
plink支持直接读取vcf文件格式,基本用法如下:
plink --vcf input.vcf --recode --out output
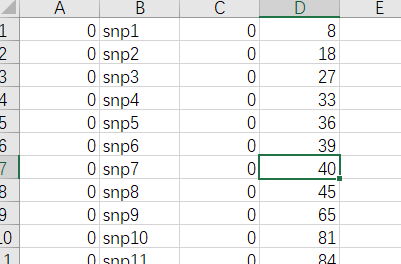

map文件 染色体编号为数字, 未知为0SNP名称为字符或数字, 如果不重要, 可以从1编号, 注意要和bed文件SNP列一一对应染色体的摩尔未知(可选项, 可以用0)SNP物理坐标
重要! 因为转换成的ped和map文件无法匹配,需要手动更改上一步转换好的map文件
map数据格式为四列:
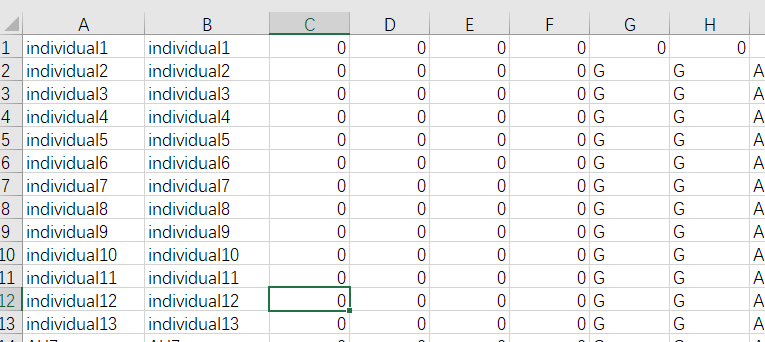

bed文件 第一列: Family ID # 如果没有, 可以用个体ID代替第二列: Individual ID # 个体ID编号第三列: Paternal ID # 父本编号第四列: Maternal ID # 母本编号第五列: Sex (1=male; 2=female; other=unknown) # 性别, 如果未知, 用0表示第六列: Phenotype # 表型数据, 如果未知, 用0表示第七列以后: 为SNP分型数据, 可以是AT CG或11 12, 或者A T C G或1 1 2 2————————————————版权声明:本文为CSDN博主「育种数据分析之放飞自我」的原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接及本声明。原文链接:https://blog.csdn.net/yijiaobani/article/details/83017730
使用plink将ped/map转换为二进制的bed文件,命令行如下:
plink --file inputfile --make-bed --out filename第一个FILENAME的后缀为.ped和.map,生成的第二个FILENAME的后缀为.bed、.bim、.fam
3. plink提取指定样本和指定SNP的数据(keep,extract函数
plink --bfile inputfile --noweb --keep sampleID.txt --recode --make-bed --out fileoutinputfile为不加.bed后缀的bed文件
其中,sampleID.txt第一列为提取的样本Family ID,第二列为Within-family ID(IID)
plink提取SNP位点:
plink --bfile file --extract snp.txt --make-bed --out snp其中,snp.txt的文件格式如下,一个SNP位点一行:
rs1
rs2
rs3
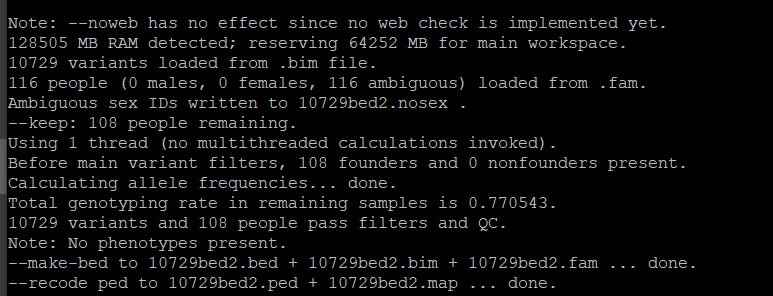
4. 如何选择合适的K值
可以同时运行多个程序, 每个程序不同的k值, 比如, 想要k值选择1,2,3,4,5, 可以写为:
for K in 1 2 3 4 5; do admixture --cv hapmap3.bed $K | tee log${K}.out; done例子:
for K in 1 2 3 4 5 6 7 8 9 10 11 12; do admixture –cv 10729bed2.bed $K | tee log${K}.out; done
多线程: admixture hapmap3.bed 3 -j 4
使用grep命令去查看*out文件的cv error(交叉验证的误差)值:
grep -h CV *.out
结果如下:(这个K值显示是否有误?应该从第一开始分别是K=1,2,3依次往下)
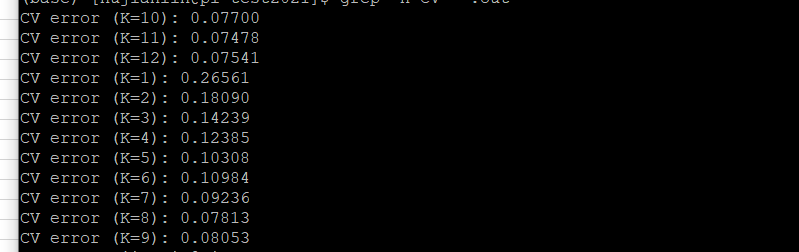
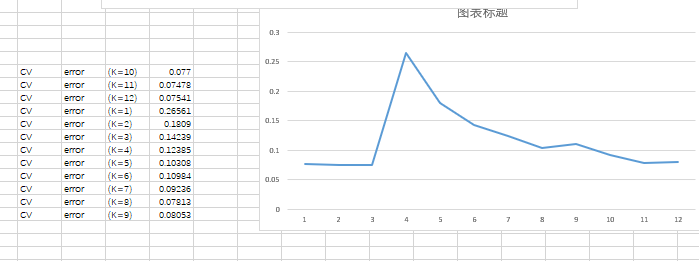
对这个K值出现这样的情况?为何K10开始,个人觉得这个K值显示有误,应该从第一开始分别是K=1,2,3依次往下
5. 绘制Q值的百分比柱状图
使用R语言
ta1 = read.table("D:/files.3.Q")
head(ta1)
barplot(t(as.matrix(ta1)),col = rainbow(3),
xlab = "Individual",
ylab = "Ancestry",
border = NA)
————————————————————————————————————————————
本文部分分析步骤参考了CSDN博主「育种数据分析之放飞自我」的原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接及本声明。
原文链接:https://blog.csdn.net/yijiaobani/article/details/83017730
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/206762.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...