大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全系列IDE使用 1年只要46元 售后保障 童叟无欺
利用R包DEseq2进行差异表达分析和可视化
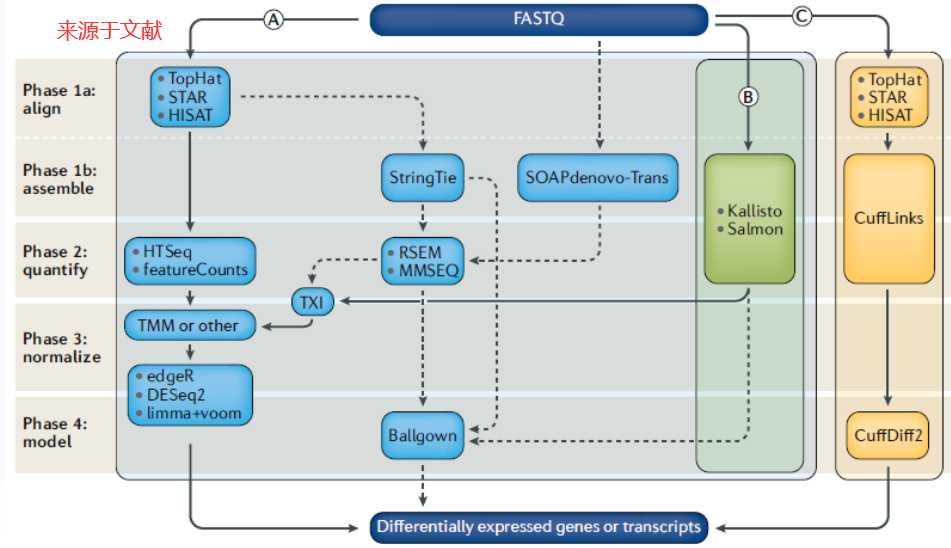
首先附上文献中的坚定差异基因的流程图。

count数矩阵
- 在Linux下,通过HISAT2 对fastq数据文件进行比对,FeatureCounts软件进行基因水平定量,得到count数矩阵。之后便可以载入R语言中进行差异分析。
差异分析
- 第一次分析RNA-seq数据,走到这一步相对容易了许多。转录组数据分析主要参考了生信技能树Jimmy老师的相关课程及推文。
- RNA-seq的read count普遍认为符合泊松分布,但是之前分析过的芯片数据符合正态分布,所以筛选DEGs的方法有一定差别。
1. 安装并载入R包
# 设置R语言镜像
# options(BioC_mirror="http://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
# options("repos" = c(CRAN="http://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
# 安装R包
# if(!require(c("ggthemes","ggpubr","ggthemes","ggrepel"))) install.packages(c("ggthemes","ggpubr","ggthemes","ggrepel"))
# BiocManager::install("DESeq2")
#载入R包
suppressPackageStartupMessages(library(DESeq2))
suppressPackageStartupMessages(library(ggpubr))
suppressPackageStartupMessages(library(ggplot2))
suppressPackageStartupMessages(library(ggrepel))
suppressPackageStartupMessages(library(ggthemes))
2. count数矩阵导入并对矩阵进行数据处理
exprset <- read.table("RNA-seq_counts_matrix.csv",sep = ",",header = T,check.names = F)
rownames(exprset) <- exprset[,1]
exprset <- exprset[,-1]
exprset <- exprset[apply(exprset,1,function(x) sum(x > 1) > 1),] #先判断值是否为0,得到逻辑值,再取和,判断0的个数是否小于3
dim(exprset)
# 7428 4
head(exprset)
head(exprset)
| control1 | control2 | treat1 | treat2 | |
|---|---|---|---|---|
| ENSMUSG00000000028 | 27 | 0 | 0 | 6 |
| ENSMUSG00000000088 | 124 | 268 | 87 | 313 |
| ENSMUSG00000000094 | 5 | 12 | 2 | 0 |
| ENSMUSG00000000131 | 17 | 5 | 6 | 5 |
| ENSMUSG00000000134 | 23 | 79 | 0 | 1 |
| ENSMUSG00000000142 | 6 | 10 | 0 | 0 |
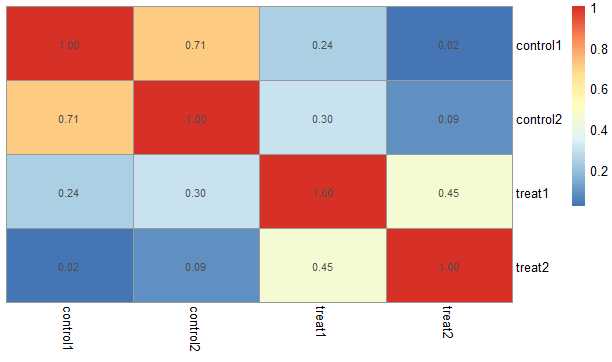
3. 查看样本相关性并采用热图展示
expcor <- cor(exprset, method = "spearman")
head(expcor)
pheatmap::pheatmap(expcor, clustering_method = "average",
treeheight_row = 0,treeheight_col = 0,
display_numbers = T)
expcor data
| control1 | control2 | treat1 | treat2 | |
|---|---|---|---|---|
| control1 | 1.0000000 | 0.7089970 | 0.2366665 | 0.0209855 |
| control2 | 0.7089970 | 1.0000000 | 0.2990182 | 0.0866515 |
| treat1 | 0.2366665 | 0.2990182 | 1.0000000 | 0.4533486 |
| treat2 | 0.0209855 | 0.0866515 | 0.4533486 | 1.0000000 |
热图展示
4. hclust对样本进行聚类分析
# t_exprset <- t(exprset)
# t_exprset <- t_exprset[,names(sort(apply(t_exprset,2,mad),decreasing = T))[1:500]]
# out.dist <- dist(t_exprset,method = 'euclidean')
# out.hclust <- hclust(out.dist,method = 'complete')
# rect.hclust(out.hclust,k=3)
# plot(out.hclust,xlab = "",main = "")
5. 构建原始dds矩阵并保存为Rdata对象
group_list <- factor(c(rep("untrt",2),rep("trt",2))) #因子型变量
group_list
table(group_list)
## group_list
## trt untrt
## 2 2
colData <- data.frame(row.names = colnames(exprset),
group_list = group_list)
colData
dds <- DESeqDataSetFromMatrix(countData = exprset,
colData = colData,
design = ~group_list) #~在R里面用于构建公式对象,~左边为因变量,右边为自变量。
head(dds)
## class: DESeqDataSet
## dim: 6 4
## metadata(1): version
## assays(1): counts
## rownames(6): ENSMUSG00000000028 ENSMUSG00000000088 ...
## ENSMUSG00000000134 ENSMUSG00000000142
## rowData names(0):
## colnames(4): control1 control2 treat1 treat2
## colData names(1): group_list
tem_f <- 'RNA-seq_DESeq2-dds.Rdata'
colData
| group_list | |
|---|---|
| control1 | untrt |
| control2 | untrt |
| treat1 | trt |
| treat2 | trt |
6. 原始dds矩阵标准化并保存
if (!file.exists(tem_f)) {
dds <- DESeq(dds) # 标准化
save(dds,file = tem_f)
}
load(file = tem_f)
# 结果用`result()`函数提取
res <- results(dds,
contrast = c("group_list","untrt","trt")) # 差异分析结果
resOrdered <- res[order(res$padj),] # 对结果按照调整后的p值进行排序
head(resOrdered)
summary(res)
##
## out of 7428 with nonzero total read count
## adjusted p-value < 0.1
## LFC > 0 (up) : 465, 6.3%
## LFC < 0 (down) : 507, 6.8%
## outliers [1] : 0, 0%
## low counts [2] : 2160, 29%
## (mean count < 4)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results
head(resOrdered)
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| ENSMUSG00000061787 | 1308.2358 | -9.456575 | 1.564545 | -6.044298 | 0e+00 | 3.60e-06 |
| ENSMUSG00000064370 | 1304.1697 | -13.689071 | 2.284209 | -5.992916 | 0e+00 | 3.60e-06 |
| ENSMUSG00000096745 | 667.1955 | -12.722138 | 2.066186 | -6.157306 | 0e+00 | 3.60e-06 |
| ENSMUSG00000096363 | 320.2598 | -11.663243 | 2.067930 | -5.640056 | 0e+00 | 2.24e-05 |
| ENSMUSG00000031504 | 229.8465 | -11.184637 | 2.077845 | -5.382805 | 1e-07 | 4.24e-05 |
| ENSMUSG00000038900 | 583.4616 | -8.543657 | 1.597311 | -5.348775 | 1e-07 | 4.24e-05 |
7. 提取差异分析的结果
DEG <- as.data.frame(resOrdered)
DESeq2_DEG <- na.omit(DEG)
diff <- subset(DESeq2_DEG,pvalue < 0.05) #先筛选P值
up <- subset(diff,log2FoldChange > 2) #上调
down <- subset(diff,log2FoldChange < -2) #下调
#可利用`write.csv()`函数保存文件
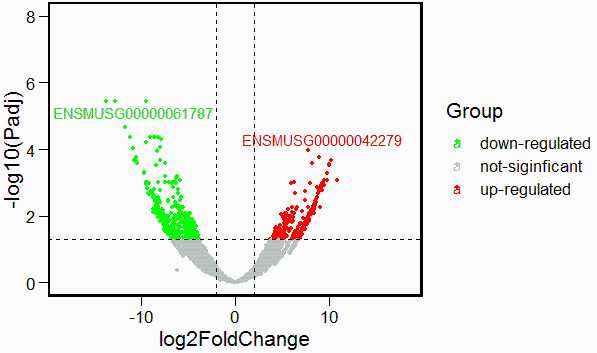
8. 绘制火山图
DEG_data <- DESeq2_DEG
DEG_data$logP <- -log10(DEG_data$padj) # 对差异基因矫正后p-value进行log10()转换
dim(DEG_data)
## [1] 5268 7
#将基因分为三类:not-siginficant,up,dowm
#将adj.P.value小于0.05,logFC大于2的基因设置为显著上调基因
#将adj.P.value小于0.05,logFC小于-2的基因设置为显著上调基因
DEG_data$Group <- "not-siginficant"
DEG_data$Group[which((DEG_data$padj < 0.05) & DEG_data$log2FoldChange > 2)] = "up-regulated"
DEG_data$Group[which((DEG_data$padj < 0.05) & DEG_data$log2FoldChange < -2)] = "down-regulated"
table(DEG_data$Group)
##
## down-regulated not-siginficant up-regulated
## 336 4659 273
DEG_data <- DEG_data[order(DEG_data$padj),]#对差异表达基因调整后的p值进行排序
#火山图中添加点(数据构建)
up_label <- head(DEG_data[DEG_data$Group == "up-regulated",],1)
down_label <- head(DEG_data[DEG_data$Group == "down-regulated",],1)
deg_label_gene <- data.frame(gene = c(rownames(up_label),rownames(down_label)),
label = c(rownames(up_label),rownames(down_label)))
DEG_data$gene <- rownames(DEG_data)
DEG_data <- merge(DEG_data,deg_label_gene,by = 'gene',all = T)
#不添加label
ggscatter(DEG_data,x = "log2FoldChange",y = "logP",
color = "Group",
palette = c("green","gray","red"),
repel = T,
ylab = "-log10(Padj)",
size = 1) +
theme_base()+
scale_y_continuous(limits = c(0,8))+
scale_x_continuous(limits = c(-18,18))+
geom_hline(yintercept = 1.3,linetype = "dashed")+
geom_vline(xintercept = c(-2,2),linetype = "dashed")

#添加特定基因label
ggscatter(DEG_data,x = "log2FoldChange",y = "logP",
color = "Group",
palette = c("green","gray","red"),
label = DEG_data$label,
repel = T,
ylab = "-log10(Padj)",
size = 1) +
theme_base()+
theme(element_line(size = 0),element_rect(size = 1.5))+ #坐标轴线条大小设置
scale_y_continuous(limits = c(0,8))+
scale_x_continuous(limits = c(-18,18))+
geom_hline(yintercept = 1.3,linetype = "dashed")+
geom_vline(xintercept = c(-2,2),linetype = "dashed")
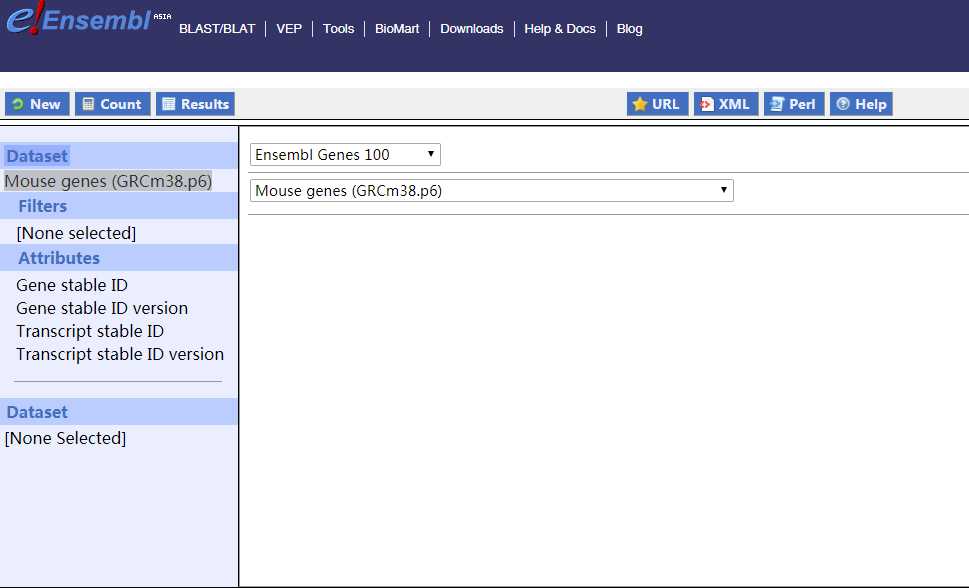
9. 简单gene ID转换
-
对于初学者来说如果要对gene ID进行转换,可利用Ensembl数据库的BioMart工具。因为相对于R包biomaRt,界面化的操作更加易懂,快捷。BioMart网页工具的原始界面如下所示:
其中左侧菜单栏分别是Dataset--选择相关物种参考基因组; Filters--选择数据gene ID的类型,并输入gene ID,也存在其他类型的ID输入; Attributes--选择需要输出的ID类型; 点击Result可以输出结果,并且支持文件下载。
- 第一次写推文,请大家多提宝贵意见!
- ##如有侵权请联系作者删除!
参考文件
[1] https://mp.weixin.qq.com/s/uDnFJC0szOHtO2NqREz2wA
[2] https://www.jianshu.com/p/3a0e1e3e41d0
[3] https://www.bioconductor.org/help/workflows/RNAseq123/
[4] https://www.bioconductor.org/help/workflows/rnaseqGene/
[5] http://www.biotrainee.com/forum.phpmod=viewthread&tid=1750#lastpost
[6] https://mp.weixin.qq.com/s/ZYB06Yudck2hD0qWJKJcwQ
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/179207.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
