大家好,又见面了,我是你们的朋友全栈君。
复合蛋白amber坐标和拓扑文件的创建
作者:朱宁 来源:大科研小分享
前言
分子动力学(Molecular Dynamics, MD)是一门结合物理,数学和化学的综合技术。目前主流分子动力学软件有NAMD、GROMACS、AMBER等。
AMBER分子动力学程序包是由加州圣弗兰西斯科大学(UCSF)的Peter A Kollman和其同事编写的,程序很全,大约包含60多个程序,相互协调工作。现在已经发展到版本20。AMBER功能涵盖种类非常多的生物分子,包括蛋白、核酸以及药物小分子。
本文中,笔者将以T4溶菌酶L99A / M102Q和2-丙基苯酚(PDB ID:3htb)的复合蛋白为例,在ubuntu环境下创建amber MD的坐标和拓扑文件,以便于学习、分享之用,如有侵权,请联系删除。
操作环境:ubuntu18.04 操作软件:ambertools19、文本编辑器 文件准备:3htb.pdb 小分子resp电荷计算网站:
https://upjv.q4md-forcefieldtools.org/REDServer-Development/
01
PDB文件的准备 首先,我们对需要模拟的复合蛋白的PDB文件进行清理 (PDB文件中用于设置amber模拟的唯一必需数据或有用数据是:原子名称,残基名称,某些与配体成键的水分子可能还有链标识符(如果存在多个链)以及重原子的坐标),对于复合蛋白如果有氢原子,建议是全部删除后,利用
reduce、H++、chimera 或者
amber 中
l
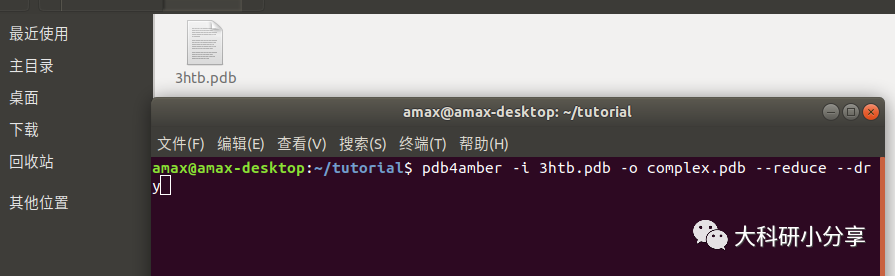
eap 添加。 打开终端,终端输入命令:
pdb4amber -i 3htb.pdb -o complex.pdb --reduce --dry # --reduce:用reduce加氢,--dry:删除所有水分子。# 如果想删除所有H原子,后面加上 -y 即可,示例中的3htb.pdb中没有H原子。注:
pdb4amber不能清理PDB文件中MD模拟不需要的链以及多余的配体,需要手动删除。同时,
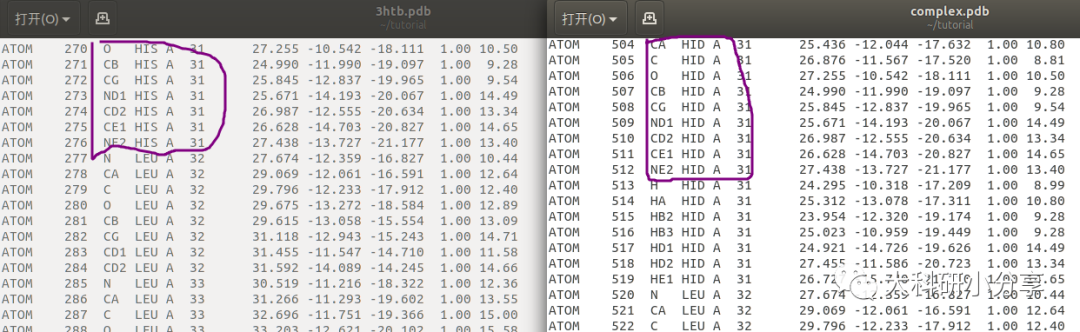
pdb4amber清理PDB文件时会预测组氨酸的质子化状态(
δ 、 ε、 δ 和ε位置均质子化),然后分别对组氨酸残基重命名(
HID,HIE,HIP)。 示例中3hth.pdb中某些组氨酸残基名
HIS,在
pdb4amber 清理PDB文件后在
complex.pdb中组氨酸残基名为
HID。对于其他残基质子化 状态可通过其他软件(
H++、PROPAK、MOE及其他药物设计软件等)进行预测,然后手动或者软件自动修改,也 有人说其他氨基酸质子化不影响H键,正好笔者比较懒,其他氨基酸质子化的步骤便省略了,但是
amber19手册 12.2有关其他氨基酸质子化残基命名的说明,因此笔者在
第四节中叙述了关于amber其他氨基酸残基的质子化状态以及命名。用
pdb4amber自动清理后,用文件编辑器打开
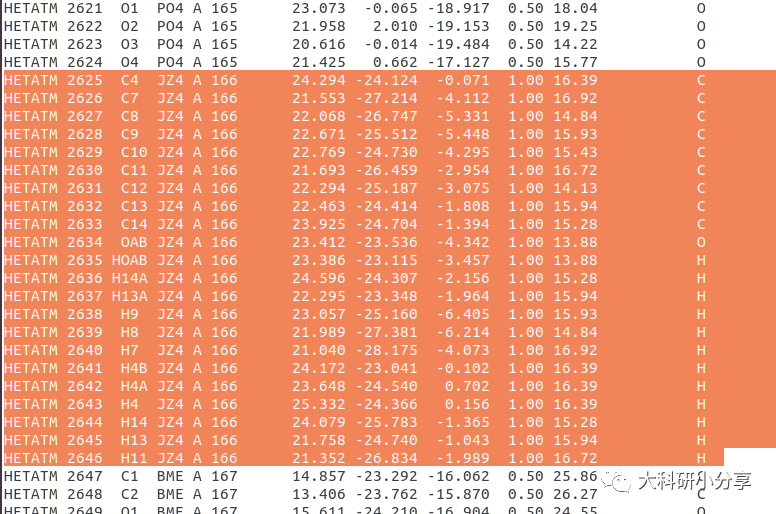
complex.pdb我们删去配体中还有多余的磷酸盐离子(
PO4)和β-巯基乙醇(
BME),保留2-丙基苯酚(
JZ4),保存
complex.pdb。删除多余的磷酸盐离子(
PO4)和β-巯基乙醇(
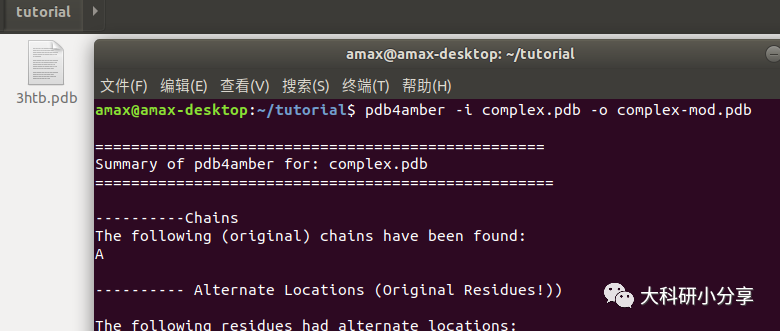
BME)后,我们对PDB文件重新编号。终端输入:
pdb4amber -i complex.pdb -o complex-mod.pdb然后复制2-丙基苯酚(
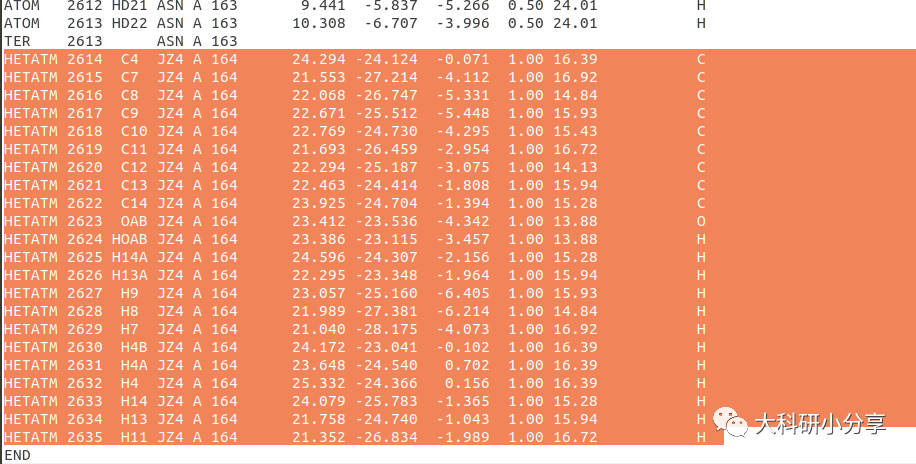
JZ4)的坐标,粘贴至另一个新建文本文件中,保存为PDB文件。这里笔者保存为
ligand.pdb,即小分子配体PDB文件。

02
小分子力场参数化小分子力场参数化主要是分两个步骤,首先我们的计算小分子的原子电荷,可以选择
bcc电荷,也可以选择
resp电荷。不要求太高精确性,就采用
bcc电荷,计算方法也简单。要求比较高的话,采用resp电荷。然后
parmchk2检查带有原子电荷的力场库文件
(
mol2 or amber prep
),补齐
gaff力场中缺失的键, 键角, 二面角参数等,生成力场参数文件
frcmod。 采用
bcc电荷的话:使用
antechamber和
parmchk2即可
antechamber -i ligand.pdb -fi pdb -o ligand.mol2 -fo mol2 -c bccparmchk2 -i ligand.mol2 -f mol2 -o ligand.frcmod 采用resp电荷的话:可以用一些量化软件(如Gaussian 、Multiwfn、GAMESS)进行计算然后拟合以及R.E.D网站。
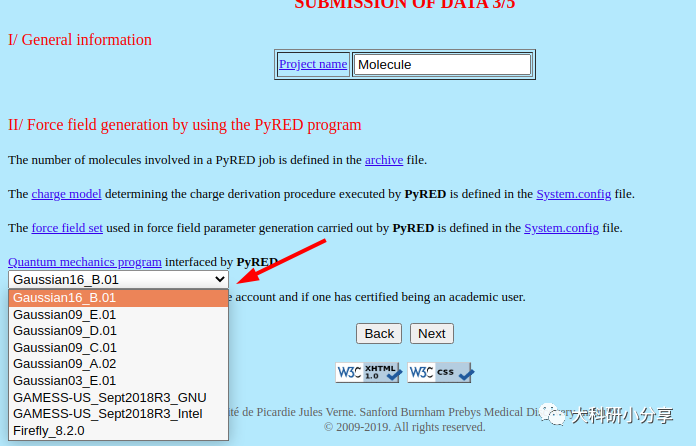
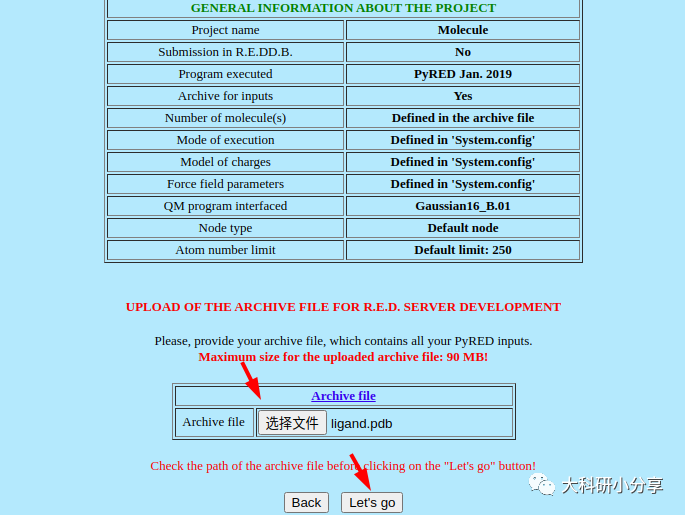
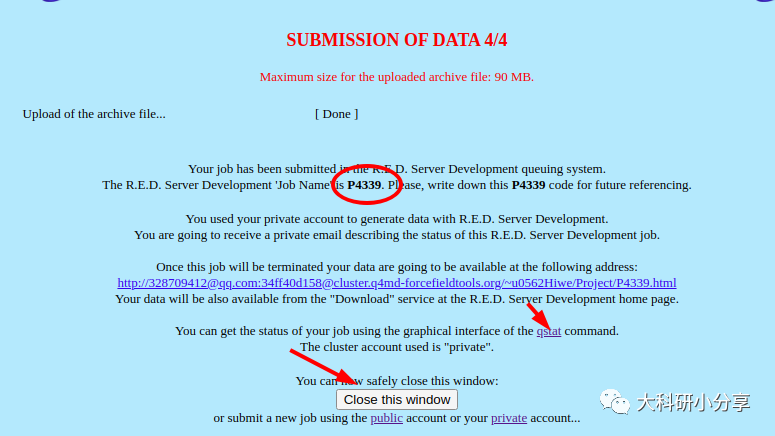
https://upjv.q4md-forcefieldtools.org/REDServer-Development/ 这里我们介绍下这个花里胡哨的网站,该网站提供电荷的计算拟合,以及力场的开发等等。当然网站是免费的,正经人谁用付费的。注册一个账号,这样网站分配的计算资源多一些,而且可以使用Gaussian计算,不用担心版权。提交文件后,他会在任务运行和结束发送邮件。使用该网站,我们必须提供的小分子的PDB文件,示例即
ligand.pdb。网站没更新前还必须提供 System.config,Project.config文件,用于自定义项目配置参数。包括分子名称,分子总电荷,自旋多重度以及拟合电荷的基组、内存、并行计算核数等。更新后就不是必要的了,我们选择默认设置就好
(
总电荷和自旋多重性分别为0和1,执行几何优化和MEP补偿、最大内存为61440 ,核心数为8等
)。如下图所示,这里我们利用网站计算生成的力场库文件
(Force field library)-Mol-sm_m1-c1.mol
2,下载文件后,移至工作目录,名字太长了,重命名 为
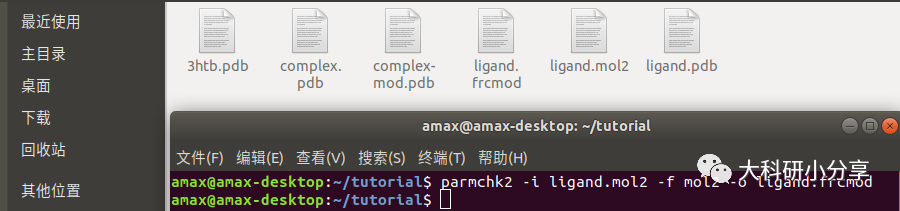
ligand.mol2 。 该网站还直接提供力场参数文件(基于amber10)和leap脚本,笔者认为其使用过于复杂,花里胡哨,故不做选择,有兴趣的可自行尝试。 获取力场库文件
ligand.mol2 ,终端输入:
parmchk2 -i ligand.mol2 -f mol2 -o ligand.frcmod这样我们就得到了力场参数文件
ligand.frcmod和以及带有原子电荷的库文件
ligand.mol2。


03
创建坐标和拓扑文件 准备好了所需的文件后,接下来终端输入以下命令即可:
tleap #打开tleapsource leaprc.protein.ff19SB #加载蛋白质力场source leaprc.gaff2 #加载gaff2力场source leaprc.water.tip3p #加载水模型loadamberparams ligand.frcmod #导入小分子力场参数文件JZ4=loadmol2 ligand.mol2 #加载小分子库文件,JZ4为PDB文件小分子残基名。com=loadpdb complex-mod.pdb #导入复合蛋白charge com #计算体系电荷,示例的体系带正电,故此加入Cl-addions com Cl- 0 #加入抗衡离子使体系电中性solvatebox com TIP3PBOX 10.0 #加入水盒子,大小为10埃saveamberparm com complex.top complex.crd #创建拓扑和坐标文件quit #退出 当然也可像笔者这样将命令放到一起,创建一个
tleap.in 的脚本,终端输入:
tleap -s -f tleap.in > tleap.out 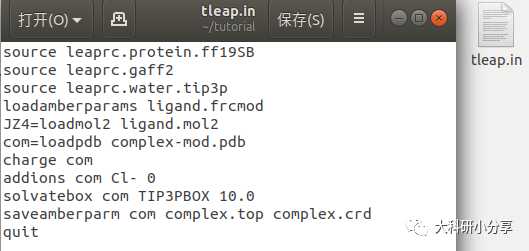
04
氨基酸残基质子化 质子化其实就是氨基酸N端多加了质子(氢原子)。氨基酸残基的质子化状态会随着蛋白质所在环境的pH值不同而发生变化,质子化的残基如果形成氢键,因为氢原子的位置不同,所以跟普通的残基也是不一样的。 对于环境pH值比较敏感的残基主要包括:HIS,ASP,GLU,LYS。 对于
HIS ,
δ 和ε位置均质子化 状态下改为
HIP ,
δ 和ε位置分别质子化为
HID 和
HIE 。
HID 和
HIE 的区别在于质子化的位置分别为
δ
N 和
ε
N ,使用
pdb4amber 时,它会自动预测并且修改。 对于
ASP,GLU,LYS质子化分别对应
ASH,GLH,LYN
。创建amber坐标和拓扑文件之前,可手动将残基名修改成对应的表示质子化的残基名,然后加氢。当然,不改对体系的影响也不大。一般情况下,修改组氨酸残基名
HIS就可以了。 最后,笔者认为如果发生质子化的残基不在活性口袋区域,那这些残基改不改都无所谓了。不在活性口袋,根本没有必要研究它,当然这仅是笔者的个人看法。 后记 对于分子动力学模拟,笔者也是刚接触不久,文中所写也是笔者自行参阅amber手册以及网上的一些资料得出,若有不当指出,欢迎指正。
 – END –
– END –
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/141360.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
