大家好,又见面了,我是你们的朋友全栈君。
利用perl一键生成符合在线LEFse差异分析的Table表
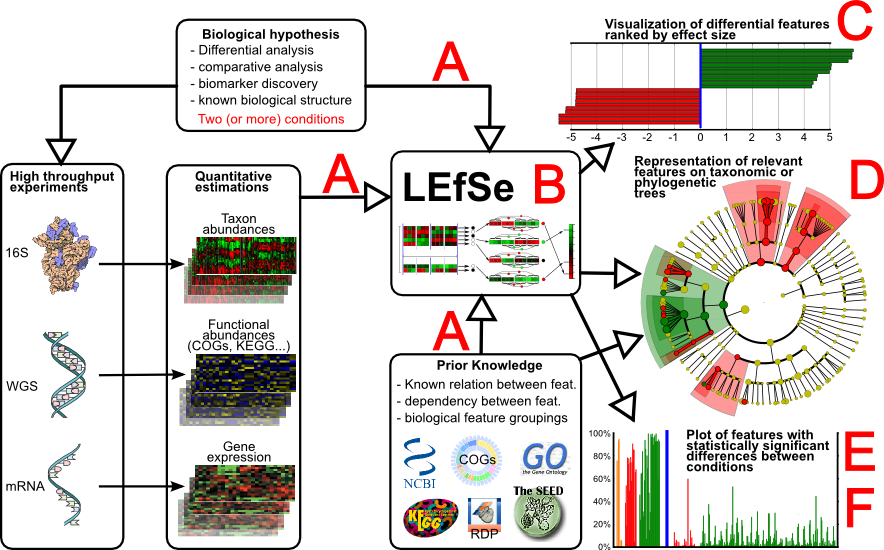
LEfSe分析的在线+本地运行的详细教程参考刘尧博客
基于Picrust2进行宏基因预测后,我们往往需要对数据进行可视化话,其中LEFse就是非常不错的选择,这里通过perl实现对表的格式化。
LEFse –Galaxy平台:http://huttenhower.sph.harvard.edu/galaxy
use strict;
use warnings;
my $mapFile=$ARGV[0];
my $tableFile=$ARGV[1];
my %mapHash=();
open(MAP,"$mapFile")or die $!;
while(my $line=<MAP>)
{
chomp($line);
my @arr=split(/\s+/,$line);
$mapHash{
$arr[0]}=$arr[3];
}
close(MAP);
my %hash=();
my @samples=();
my %sumHash=();
open(RF,"$tableFile") or die $!;
while(my $line=<RF>)
{
next if($.==1);
my @arr=split(/\t/,$line);
if($.==2)
{
@samples=@arr;
next;
}
my $taxonomy=$arr[$#arr];
$taxonomy=~s/;/|/g;
$taxonomy=~s/(k|p|c|o|f|g|s)__//g;
$taxonomy=~s/\s+|\[|\]//g;
$taxonomy=~s/\|*$//g;
my @taxoArr=split(/\|/,$taxonomy);
my $taxSetp="";
for(my $tax=0;$tax<=$#taxoArr;$tax++)
{
if($tax==0)
{
$taxSetp=$taxoArr[$tax];
}
else
{
$taxSetp=$taxSetp . "|" . $taxoArr[$tax];
}
for(my $i=1;$i<$#samples;$i++)
{
${
$hash{
$taxSetp}}{
$samples[$i]}+=$arr[$i];
}
}
for(my $i=1;$i<$#samples;$i++)
{
$sumHash{
$samples[$i]}+=$arr[$i];
}
}
close(RF);
open(WF,">lefse_input.txt")or die $!;
#print WF "Taxonomy";
#for(my $i=1;$i<$#samples;$i++)
#{
# print WF "\t" .$samples[$i];
#}
#print WF"\n";
print WF "sampleType";
for(my $i=1;$i<$#samples;$i++)
{
print WF "\t" . $mapHash{
$samples[$i]};
}
print WF "\n";
foreach my $key(sort (keys %hash))
{
next if($key=~/Unassigned/);
print WF $key;
for(my $i=1;$i<$#samples;$i++)
{
print WF "\t" . ${
$hash{
$key}}{
$samples[$i]}/$sumHash{
$samples[$i]}*100;
}
print WF "\n";
}
close(WF);
将以上代码保存为lefse.pl, 然后cd到相应目录,将输入文件lefse_input.txt(以picrust2输出的meta表为例) ,和map文件 sample-meta.txt,放到该pl同一目录下,然后运行
perl lefse.pl
然后就可以根据官方教程,进行上传生成LEFse分析表格。

更多微生物组学分析代码与方法,欢迎访问zlab仓库https://github.com/zlabx
参考文献:
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 举报,一经查实,本站将立刻删除。
发布者:全栈程序员-用户IM,转载请注明出处:https://javaforall.cn/136569.html原文链接:https://javaforall.cn
【正版授权,激活自己账号】: Jetbrains全家桶Ide使用,1年售后保障,每天仅需1毛
【官方授权 正版激活】: 官方授权 正版激活 支持Jetbrains家族下所有IDE 使用个人JB账号...
